 手機版
手機版
 關于我們
關于我們 加入收藏
加入收藏

步琦實驗室設備貿易(上海)有限公司
3 年 白金會員
白金會員
白金會員 已認證
已認證
撥打電話
獲取底價
提交后,商家將派代表為您專人服務
步琦實驗室設備貿易(上海)有限公司
白金會員已認證

利用步琦SFC系統純化
阿司匹林、苯佐卡因和普魯卡因
SFC應用
1
簡介
化學合成常伴有雜質,因產率很少達 100%,雜質會嚴重影響藥物療效、安全和質量,所以純化藥物保障其純度和完整性至關重要,在現代藥物研發的過程中往往通過色譜法等多種方法純化。
在上一篇應用文章《利用步琦 SFC 系統純化利多卡因與乙酰氨基酚》中,我們使用超臨界流體色譜(SFC)高效純化了利多卡因和乙酰氨基酚的合成產物。在此過程中,我們先使用步琦獨有的 Sepmatix 8x SFC 平行色譜系統快速篩選了色譜柱,之后將其放大到 BUCHI Sepiatec SFC-50 超臨界制備色譜系統上。本次應用中,我們依舊會使用同樣的工作流程去純化阿司匹林、苯佐卡因和普魯卡因的合成產物。
2
實驗設備
BUCHI Sepmatix 8x SFC 8通道平行色譜系統
BUCHI Sepiatec SFC-50 超臨界制備色譜系統
BUCHI PrepPure 硅膠,5um,250×4.6mm
BUCHI PrepPure 二醇基,5um,250×4.6mm
BUCHI PrepPure 氨基,5um,250×4.6mm
BUCHI PrepPure 2-EP,5um,250×4.6mm
HILIC柱,5um,250×4.6mm (Dr. Maisch GmbH)
BUCHI PrepPure PEI,5um,250×4.6mm
BUCHI PrepPure PEI,5um,250×4.6mm
BUCHI PrepPure CBD,5um,250×4.6mm
氰基柱,5um,250×10mm ,(Dr. Maisch GmbH)
BUCHI PrepPure 2-EP,5um,250×10mm
BUCHI PrepPure 二醇基,5um,250×10mm
3
化學品與樣品
化學品:
二氧化碳 (99.9%)
甲醇 (≥99%)
甲醇溶液中 2M 的氨溶液
甲酸(99%)
去離子水
為了安全處理,請注意所有相應的MSDS!
樣品:
普魯卡因合成產物
阿司匹林合成產物
苯佐卡因合成產物
4
程序設定
BUCHI Sepmatix 8x SFC 平行色譜柱系統
流動相:A= 二氧化碳;B= 甲醇
柱尺寸:250×4.6mm
流速:3mL/min(每根色譜柱)
檢測:DAD 紫外掃描 200 nm - 600 nm
流動相條件:
0 – 0.5 min | 5 % B | |
0.5 – 8.0 min | 5 – 50 % B | |
8.0 – 9.4 min | 50 % B | |
9.4 – 9.5 min | 50 – 5 % B | |
9.5 – 10 min | 5 % B | |
篩選運行完全自動運行,流速設置為 3mL/min 每通道,使用流控單元,平衡每一根色譜柱。樣品自動注入(V=5μL),并開始平行篩選(運行時間=10min)。背壓調節器設置為 150 bar,柱子加熱至 32℃,可按需往改性劑中加入添加劑改善峰型。
BUCHI Sepiatec SFC-50 超臨界制備色譜系統
流動相:A= 二氧化碳;B= 甲醇
柱尺寸:250×10mm
流動相條件:等度運行條件
檢測:紫外
所有 10mm ID 色譜柱都在預設流速下平衡 3 分鐘,使用自動進樣器上樣,并開始運行。背壓調節器設置為 150 bar,柱子加熱至 40℃,可按需往改性劑中加入添加劑改善峰型。
5
結果
5.1 阿司匹林
阿司匹林,化學名為乙酰水楊酸(下稱 ASA),是一種眾所周知的鎮痛和抗炎藥物。它屬于非甾體抗炎藥(NSAIDs)這一類。可以通過將水楊酸(下稱 SA)與乙酸酐發生酯化反應而化學合成(見圖5) [1] 。為了確定乙酰水楊酸的理想分離選擇性,首先進行了色譜柱柱篩選(見圖1)。
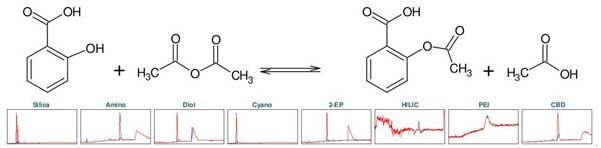
▲ 圖 1:頂部:阿司匹林合成的反應方程式,底部:Sepmatix 8x SFC 儀器色譜柱篩選結果;從左到右:硅膠,氨基,二醇基,氰基,2-EP,HILIC,PEI和CBD;運行時間 = 10分鐘。
由于 SA 在固定相中的保留性較高,為了強制洗脫 SA,在梯度中增加了一個等度步驟,即在 50% 改性劑濃度下進行 5 分鐘的等度洗脫。在 HILIC 相中,ASA 和 SA 只有噪音而無洗脫。在 PEI 相中,在實驗條件下只有 ASA 被洗脫。除氰基相外,其余各相均顯示出 ASA 和 SA 的分離。結合分離時長和分辨率而言,2-EP 相的結果最好(見表 1)
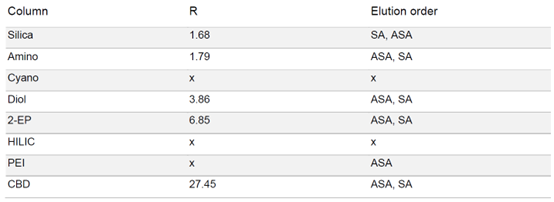
▲ 表1:樣品在不同固定相色譜柱條件下的分辨率值和洗脫順序
選擇 2-EP 相作為固定相,放大至制備規模。在色譜柱篩選的過程中,ASA 和 SA 在 2-EP上被洗脫的甲醇比例為 26 - 40%。圖 2(上)顯示了在甲醇比例為 40% 的10x250mm 2-EP 色譜柱上純化 ASA 和 SA 的情況。改性劑中添加了甲酸(0.5%),以改善 SA 的峰形。在不添加甲酸的情況下,SA 的拖尾非常嚴重。在相同條件下,可采用疊加進樣法自動純化 ASA(見圖2下),并進行餾分收集。
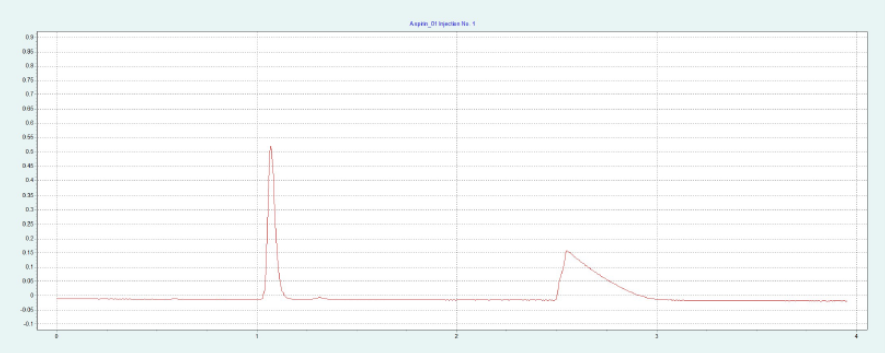
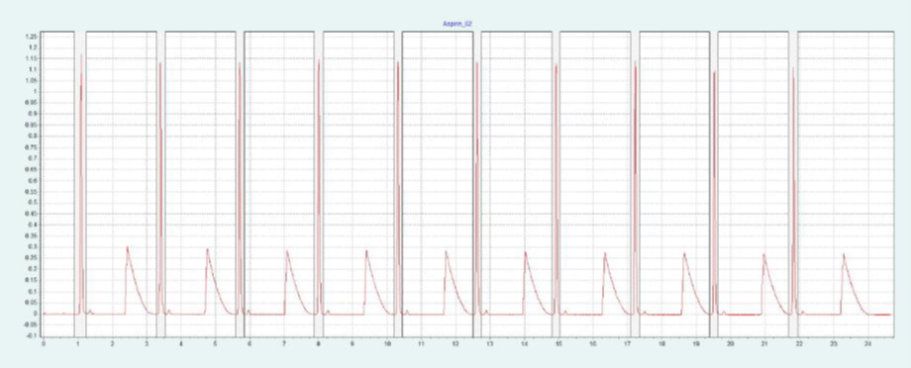
▲ 圖 2:ASA 純化的單次進樣(上圖)和疊加進樣(下圖);運行條件:流速 = 30 mL/min,改性劑 = 甲醇 + 0.5 % 甲酸,改性劑 % = 40 %,溫度 = 40 °C,壓力 BPR = 150 bar,進樣量 = 200 μL,紫外波長 = 254 nm;疊加進樣條件:進樣次數 = 10,疊加時間 = 2.0 min,餾分 = 1(基于時間)。
5.2 苯佐卡因
苯佐卡因(下稱 BC),化學名為對氨基苯甲酸乙酯,屬于局部麻醉劑類。它可以從對氨基苯甲酸(下稱 PABA)通過酸催化羧基與乙醇的酯化反應化學合成,用硫酸作為催化劑 [2]。首先進行色譜柱篩選,以確定苯佐卡因合成產物理想的分離選擇性(見 圖3)。
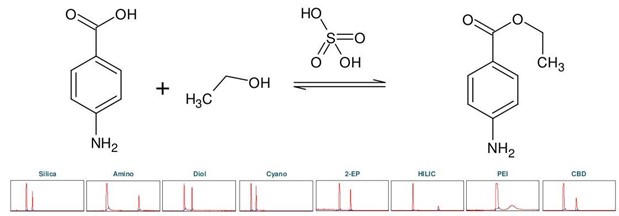
▲ 圖3:頂部:苯佐卡因合成反應方程式,底部:Sepmatix 8x SFC儀器色譜柱篩選結果;從左到右:硅膠,氨基,二醇基,氰基,2-EP,HILIC,PEI和CBD;運行時間 = 15分鐘。
由于 PABA 在固定相中的保留性較強,因此在梯度中增加了一個等度步驟,在 50% 甲醇的條件下運行 5 分鐘,硫酸通過洗滌步驟去除避免過高的酸性。各固定相的色譜圖顯示,每種固定相都能分離苯佐卡因,但是洗脫速度和分辨率存在很大差異(見表2)。HILIC 和氨基相的分辨率最高,但 PABA 的保留時間過長,即使用50%的甲醇洗脫,依舊需要很長時間。
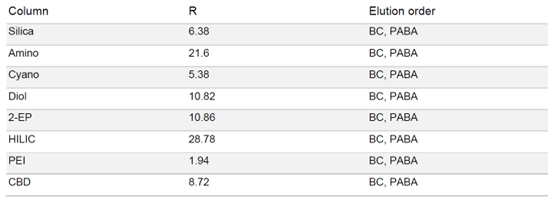
▲ 表2:樣品在不同固定相色譜柱條件下的分辨率值和洗脫順序。
結合柱篩選結果,最終選擇二醇基相作為制備色譜柱。HILIC 和氨基相需要較高比例的甲醇和較長的運行時間。二醇基相不僅可以非常快速地洗脫樣品,并具有足夠高的分辨率。二醇基柱的篩選結果表明,BC 和 PABA 在甲醇比例為 30 - 40% 時即可洗脫。
圖 4(上圖)顯示了 10 x 250mm 的二醇基色譜柱在甲醇比例為 28% 時對苯佐卡因的純化情況。在相同條件下,可采用疊加進樣法自動純化 BC(見圖4下),并進行餾分收集。
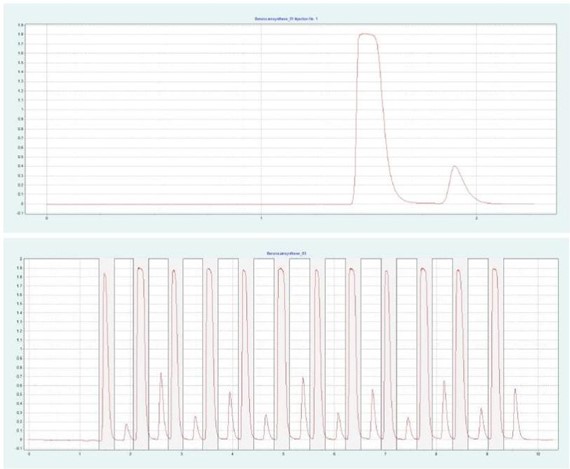
▲ 圖 4:苯佐卡因的單次注射(頂部)和堆疊注射(底部)純化;運行條件:流速 = 20 mL/min, 改性劑 = 甲醇,改性劑 % = 28%,溫度 = 40 °C,壓力 BPR = 150 bar,注射量 = 90 μL,UV 波長 =276nm;堆疊注射條件:注射次數 =12,堆疊時間 =0.66min,餾分 =1(基于時間的)。
5.3 普魯卡因
普魯卡因(下稱 PC),化學名為 4-氨基苯甲酸 2-(N, N-二乙基氨基)乙酯,是一種藥物,具有局部麻醉作用。它阻斷電壓依賴性鈉離子通道,從而導致例如疼痛敏感性的降低。普魯卡因可以通過化學方法在堿性條件下從 4-氨基苯甲酸乙酯(下稱ABE)合成。這涉及到使用 2-二乙基氨基乙醇進行酯交換[3]。堿是乙醇鈉醇,它可以通過鈉與乙醇的反應產生。為確定普魯卡因合成產物純化的理想選擇性,進行了色譜柱篩選(見圖 5)。
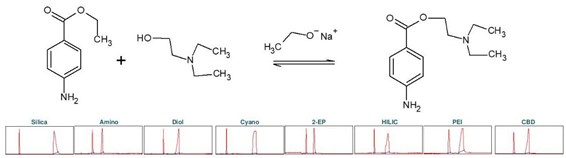
▲ 圖5:頂部:普魯卡因合成的反應方程式,底部:Sepmatix 8x SFC 儀器色譜柱篩選結果;從左到右:硅膠,氨基,二醇基,氰基,2-EP,HILIC,PEI 和 CBD;運行時間 = 15分鐘。
由于普魯卡因在固定相中的保留性較高,因此在梯度中增加了一個等度步驟,即在 50% 甲醇濃度下進行 5 分鐘的梯度。結果顯示,每種固定相對普魯卡因的純化都有合適的選擇性(見表3)。普魯卡因在硅膠、PEI 和氰基固定相中的保留性較高。
由于普魯卡因的基本特性,在部分色譜柱上的峰形非常不對稱。二醇基相下的峰前沿非常明顯,不對稱度系數為 0.58,而硅膠相下的拖尾非常明顯,不對稱度系數為 2.73。
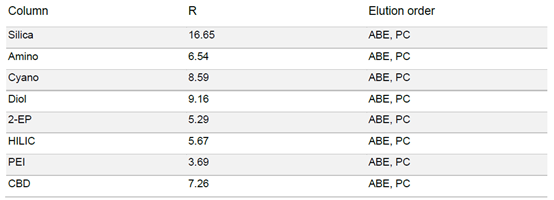
▲ 表3:樣品在不同固定相色譜柱條件下的分辨率值和洗脫順序。
結合柱篩選結果,選擇 2-EP 相放大為制備純化的方法。硅膠相需要高含量的甲醇才能洗脫出 PC,導致運行時間長,峰形不佳。而 2-EP 相既可以快速洗脫樣品,又具有足夠高的分辨率。為了改善峰型,在甲醇改性劑中加入 5 % 的去離子水和 20 mM 的氨水作為添加劑。
篩選結果表明,PC 和 ABE 在改性劑含量約為 24 - 32% 時可以從色譜柱內洗脫下來。圖6(上圖)顯示了10 x 250mm 的 2-EP 色譜柱在改性劑含量為 25% 時純化 PC 的情況。在相同條件下,可采用疊加進樣法自動純化 PC(見圖6,下圖)并收集餾分,添加添加劑可顯著改善 PC 的峰形。
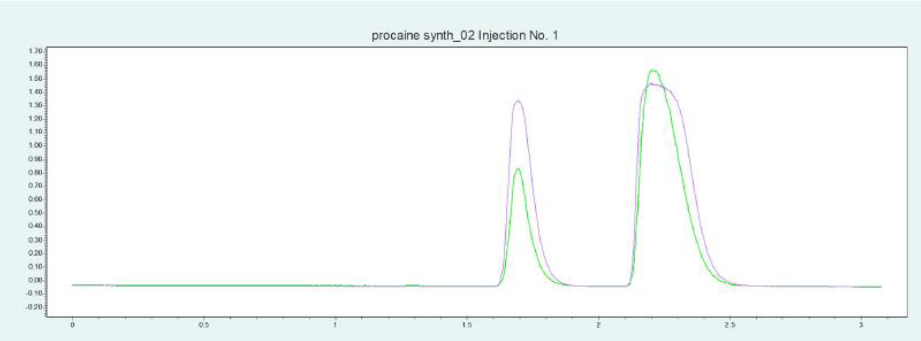
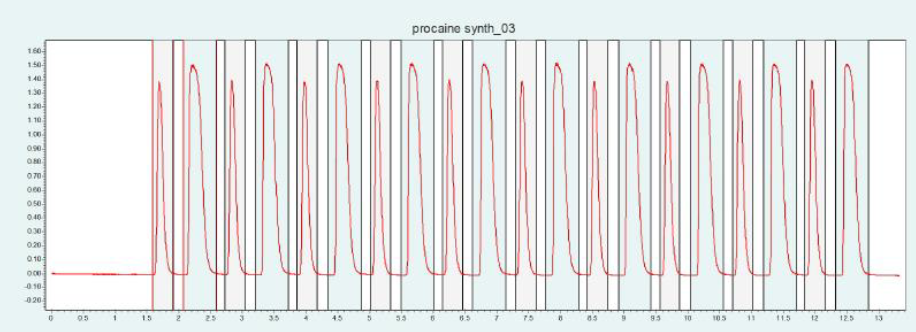
▲ 圖 6:普魯卡因純化的單次進樣(上圖)和疊加進樣(下圖);運行條件:流速 = 20 mL/min,改性劑 = 甲醇/水(95/5 %)加 20 mM 氨水,改性劑 % = 25 %,溫度 = 40 °C,壓力 BPR = 150 bar,進樣量 = 100 μL,紫外波長 = 220 nm;疊加進樣條件:進樣次數 = 10,疊加時間 = 1.1min,餾分 = 2(基于時間)。
6
結論
超臨界制備色譜純化合成產物高效快速,但需篩選合適的色譜填料。BUCHI Sepmatix 8x SFC 平行色譜系統可快速篩選填料并放大至制備規格,既能提高樣品分離度,又能充分利用色譜柱上樣量,為 BUCHI Sepiatec SFC-50 制備系統的疊加進樣奠定良好基礎,二者結合可達到降本增效的目的。
7
參考文獻
Th. Eicher und H. J. Roth; Synthese, Gewinnung und Charakterisierung von Arzneistoffen, Georg Thieme Verlag, Stuttgart (1986).
Winterfeld, K. – Praktikum der organisch-pr?parativen Pharmazeutischen Chemie, 6. Auflage, Steinkopff Verl., Darmstadt (1965).
Axel Kleemann, Jürgen Engel, Bernd Kutscher und Dietmar Reichert: Pharmaceutical Substances, 4. Auflage, Georg Thieme Verlag, Stuttgart (2000).

長按上方二維碼聯系我們
或撥打聯系電話:
400 - 880 - 8720
微信公眾號
步琦智慧實驗室
淘寶官方旗艦店
瑞士步琦
最新動態
更多






虛擬號將在 秒后失效
使用微信掃碼撥號


 上一篇
上一篇